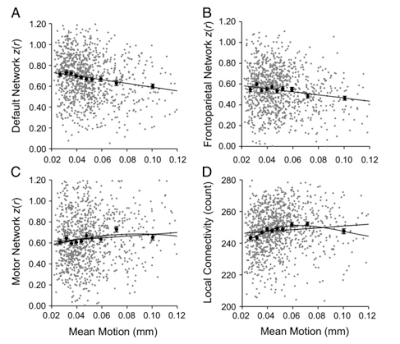
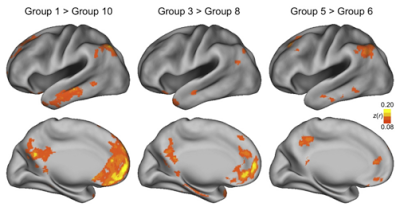
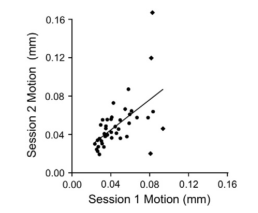
Once 3dReHo is completed and you have your correlation maps, you will then need to normalize them. Note that normalize can have different meanings, depending on the context. With FMRI data, when we talk about normalization we usually mean normalizing the data to a standard space so that individual subjects, with all of their weird-looking, messed-up brains, can be compared to each other. This results in each subject's brain conforming to the same dimensions and roughly the same cortical topography as a template, or "normal" brain, which in turn is based on the brain of an admired and well-loved public figure, such as 50 Cent or Madonna.
However, normalization can also refer to altering the signal in each voxel to become more normally distributed. This makes the resulting statistical estimates - whether betas, correlation coefficients, or z-scores - more amenable for a higher-level statistical test that assumes a normal distribution of scores, such as a t-test. Although there are a few different ways to do this, one popular method in papers using ReHo is to take the mean Kendall's Correlation Coefficient (KCC) of all voxels in the whole-brain mask, subtract that mean from each voxel, and divide by the KCC standard deviation across all voxels in the whole-brain mask.
As a brief intermezzo before going over how to do this in AFNI, let us talk about the difference between 3d and 1d tools, since knowing what question you want to answer will direct you toward which tool is most appropriate for the job. 3d tools - identified by their "3d" prefix on certain commands, such as 3dTfitter or 3dDeconvolve - take three-dimensional data for their input, which can be any three-dimensional dataset of voxels, and in general any functional dataset can be used with these commands. Furthermore, these datasets usually have a temporal component - that is, a time dimension that is the length of however many images are in that dataset. For example, a typical functional dataset may have several voxels in the x-, y-, and z-directions, making it a three-dimensional dataset, as well as a time component of one hundred or so timepoints, one for each scan that was collected and concatenated onto the dataset.
1d tools, on the other hand, use one-dimensional input, which only goes along a single dimension. The ".1D" files that you often see input to 3dDeconvolve, for example, have only one dimension, which usually happens to be time - one data point for every time point. Since we will be extracting data from a three-dimensional dataset, but only one value per voxel, then, we will use both 3d and 1d tools for normalization.
Once we have our regional homogeneity dataset, we extract the data with 3dmaskdump, omitting any information that we don't need, such as the coordinates of each voxel using the -noijk option:
3dmaskdump -noijk -mask mask_group+tlrc ReHo_Test_Mask+tlrc > ReHo_Kendall.1D
Note also that the -mask option is using the whole-brain mask, as we are only interested in voxels inside the brain for normalization.
The next step will feed this information into 1d_tool.py, which calculates the mean and standard deviation of the Kendall correlation coefficients across all of the voxels:
1d_tool.py -show_mmms -infile ReHo_Kendall.1D
Alternatively, you can combine both of these into a single using the "|" character will feed the standard output from 3dmaskdump into 1d_tool.py:
3dmaskdump -noijk -mask mask_group+tlrc ReHo_Test_Mask+tlrc | 1d_tool.py -show_mmms -infile -
Where the "-" after the -infile option means to use the standard output of the previous command.
3dcalc is then used to create the normalized map:
3dcalc -a ReHo_Test_Mask+tlrc -b mask_group+tlrc -expr '((a-0.5685)/0.1383*b)' -prefix ReHo_Normalized
Lastly, although we have used the whole brain for our normalization, a strong argument can be made for only looking at voxels within the grey matter by masking out the grey matter, white matter, and cerebrospinal fluid (CSF) separately using FreeSurfer or a tool such as 3dSeg. The reason is that the white matter will tend to have lower values for Kendall's correlation coefficient, and grey matter will have relatively higher values; thus, normalizing will bias the grey matter to have higher values than the grey matter and CSF. However, one could counter that when a second-level test is carried out across subjects by subtracting one group from another, it is the relative differences that matter, not the absolute number derived from the normalization step.
In sum, I would recommend using a grey mask, especially if you are considering lumping everyone together and only doing a one-sample t-test, which doesn't take into account any differences between groups. As with most things in FMRI analysis, I will leave that up to your own deranged judgment.
However, normalization can also refer to altering the signal in each voxel to become more normally distributed. This makes the resulting statistical estimates - whether betas, correlation coefficients, or z-scores - more amenable for a higher-level statistical test that assumes a normal distribution of scores, such as a t-test. Although there are a few different ways to do this, one popular method in papers using ReHo is to take the mean Kendall's Correlation Coefficient (KCC) of all voxels in the whole-brain mask, subtract that mean from each voxel, and divide by the KCC standard deviation across all voxels in the whole-brain mask.
As a brief intermezzo before going over how to do this in AFNI, let us talk about the difference between 3d and 1d tools, since knowing what question you want to answer will direct you toward which tool is most appropriate for the job. 3d tools - identified by their "3d" prefix on certain commands, such as 3dTfitter or 3dDeconvolve - take three-dimensional data for their input, which can be any three-dimensional dataset of voxels, and in general any functional dataset can be used with these commands. Furthermore, these datasets usually have a temporal component - that is, a time dimension that is the length of however many images are in that dataset. For example, a typical functional dataset may have several voxels in the x-, y-, and z-directions, making it a three-dimensional dataset, as well as a time component of one hundred or so timepoints, one for each scan that was collected and concatenated onto the dataset.
1d tools, on the other hand, use one-dimensional input, which only goes along a single dimension. The ".1D" files that you often see input to 3dDeconvolve, for example, have only one dimension, which usually happens to be time - one data point for every time point. Since we will be extracting data from a three-dimensional dataset, but only one value per voxel, then, we will use both 3d and 1d tools for normalization.
Once we have our regional homogeneity dataset, we extract the data with 3dmaskdump, omitting any information that we don't need, such as the coordinates of each voxel using the -noijk option:
3dmaskdump -noijk -mask mask_group+tlrc ReHo_Test_Mask+tlrc > ReHo_Kendall.1D
Note also that the -mask option is using the whole-brain mask, as we are only interested in voxels inside the brain for normalization.
The next step will feed this information into 1d_tool.py, which calculates the mean and standard deviation of the Kendall correlation coefficients across all of the voxels:
1d_tool.py -show_mmms -infile ReHo_Kendall.1D
Alternatively, you can combine both of these into a single using the "|" character will feed the standard output from 3dmaskdump into 1d_tool.py:
3dmaskdump -noijk -mask mask_group+tlrc ReHo_Test_Mask+tlrc | 1d_tool.py -show_mmms -infile -
Where the "-" after the -infile option means to use the standard output of the previous command.
3dcalc is then used to create the normalized map:
3dcalc -a ReHo_Test_Mask+tlrc -b mask_group+tlrc -expr '((a-0.5685)/0.1383*b)' -prefix ReHo_Normalized
Lastly, although we have used the whole brain for our normalization, a strong argument can be made for only looking at voxels within the grey matter by masking out the grey matter, white matter, and cerebrospinal fluid (CSF) separately using FreeSurfer or a tool such as 3dSeg. The reason is that the white matter will tend to have lower values for Kendall's correlation coefficient, and grey matter will have relatively higher values; thus, normalizing will bias the grey matter to have higher values than the grey matter and CSF. However, one could counter that when a second-level test is carried out across subjects by subtracting one group from another, it is the relative differences that matter, not the absolute number derived from the normalization step.
In sum, I would recommend using a grey mask, especially if you are considering lumping everyone together and only doing a one-sample t-test, which doesn't take into account any differences between groups. As with most things in FMRI analysis, I will leave that up to your own deranged judgment.